Parkinson vs Huntington Disease: Symptoms, Diagnosis, Care
Apr 22, 2026

A family arrives worried about a new tremor in a retired parent. On the same day, another family asks why a middle-aged sibling has become more irritable, more impulsive, and oddly fidgety in ways they can’t explain. Both conversations may lead to the same broad question: is this Parkinson’s disease, Huntington’s disease, or something else entirely?
That’s where the confusion often begins. Both conditions affect movement. Both can affect thinking, mood, and day-to-day independence. Both involve the basal ganglia. But in practice, parkinson vs huntington disease isn’t a simple tremor-versus-chorea comparison. The more useful distinction often comes from pattern recognition across age of onset, family history, psychiatric change, cognitive profile, and the quality of motor control over time.
Clinicians, therapists, and families usually need more than a label. They need to know what signs deserve urgency, what treatment helps one condition but harms the other, and what kinds of cognitive monitoring are worth doing early. That broader framework is also central to understanding degenerative diseases, especially when care planning has to bridge neurology, rehabilitation, mental health, and family support.
Distinguishing Two Faces of Neurodegeneration
A useful starting point is the bedside impression.
One person, usually older, describes moving more slowly. Their spouse notices less arm swing, a softer voice, trouble getting started when walking, and a faint resting tremor. Another person, often younger, presents with clumsiness that looks almost careless at first. They miss steps, seem unable to keep still, and family members often mention irritability or personality change before anyone mentions a movement disorder.
Those early impressions matter because they shape the next questions. In the first case, the examiner often looks for bradykinesia, rigidity, and a classic parkinsonian gait. In the second, the examiner should ask harder questions about family history, behavioural change, executive decline, and involuntary movements that are not rhythmic but flowing, fragmented, and unpredictable.
Feature | Parkinson’s disease | Huntington’s disease |
|---|---|---|
Typical onset | Usually after age 60 | Often between ages 30 and 55 |
Sex pattern | More common in males | Affects males and females equally |
Core motor pattern | Slowness, rigidity, resting tremor | Chorea, motor impersistence, instability |
Cognitive pattern | Early executive and processing speed changes | Earlier broader decline involving memory and fluency |
Psychiatric pattern | Depression, anxiety, apathy can occur | Irritability, depression, anxiety, behavioural disruption often more prominent |
Diagnostic anchor | Clinical diagnosis supported by examination and response pattern | Genetic inheritance pattern and confirmatory genetic testing |
In clinic, families often ask why these disorders get mixed up at all. The answer is that early disease rarely reads like a textbook. Parkinson’s can begin without obvious tremor. Huntington’s can begin with mood change, poor judgement, or subtle restlessness rather than dramatic chorea. A patient may also underreport symptoms, while relatives describe a very different picture.
Practical rule: If the movement complaint seems mild but the behavioural or executive changes are out of proportion, Huntington’s needs to stay on the differential.
The distinction becomes clearer once we look beneath the motor surface and examine where each disease starts, how it spreads, and what it tends to disrupt first.
Unpacking the Origins Etiology and Pathophysiology
Parkinson’s disease and Huntington’s disease both affect circuits that help organise movement, cognition, and behaviour. But they do not begin the same way, and that difference explains much of what follows clinically.
Nationally, Parkinson’s disease affects approximately 500,000 to 1 million Americans, with around 60,000 new cases annually, whereas Huntington’s disease affects about 15,000 people nationwide according to this comparison of Parkinson’s and Huntington’s disease. The same source notes that Parkinson’s typically begins after age 60 and is 1.5 times more common in males, while Huntington’s usually begins between ages 30 and 55 and affects males and females equally because of its inherited genetic basis.
Parkinson’s as a dopamine production problem
In Parkinson’s disease, the simplest clinical model is a dopamine production failure. Neurons involved in dopamine signalling, especially in the substantia nigra, progressively fail. The downstream effect is impaired signalling through basal ganglia circuits that normally help initiate and scale movement.
That’s why the syndrome often looks hypokinetic. Patients don’t just move less. They struggle to generate movement efficiently. Their gait shortens, facial expression reduces, handwriting shrinks, and automatic movements become effortful. The chemistry matters because many treatment decisions in Parkinson’s are built around restoring or mimicking dopaminergic function.
Huntington’s as a faulty genetic blueprint
Huntington’s disease follows a different logic. It is a genetic neurodegenerative disorder linked to a mutated HTT gene. In clinical terms, that means the disease isn’t primarily a neurotransmitter shortage. It is an inherited blueprint problem that leads to progressive neuronal dysfunction and loss, particularly in striatal circuits.
The result is not only abnormal movement but also earlier disturbance in behavioural regulation, impulse control, and higher-order cognition. Families often sense that “something is off” before a clinician can comfortably name the syndrome. That intuition is often correct.
For readers who want a quick refresher on how different brain systems contribute to behaviour and cognition, this guide to the lobes du cerveau is a helpful companion when explaining symptoms to families in plain language.
Why the biology changes the clinical mindset
The practical consequence is that these are not two versions of the same disease. One is usually a later-onset disorder with prominent dopaminergic dysfunction and classically slowed movement. The other is a hereditary disorder with a broader behavioural and cognitive footprint that often arrives earlier in adult life.
That distinction changes how we talk to families:
For suspected Parkinson’s disease, clinicians usually explore symptom evolution, medication response, gait pattern, and associated non-motor changes.
For suspected Huntington’s disease, clinicians need a sharper focus on family history, psychiatric symptoms, work decline, impulsivity, and the emotional implications of genetic risk.
For both, it helps to explain that the basal ganglia influence much more than movement. They shape timing, inhibition, planning, and behavioural control.
A useful analogy for families is this: Parkinson’s often begins like a system running low on the chemical fuel needed for smooth movement. Huntington’s begins like a corrupted instruction set affecting movement, thinking, and behaviour at the same time.
When teams understand that difference early, the later findings make much more sense.
Comparing Motor Symptoms Tremors Versus Chorea
The motor contrast is where most families begin, but it’s also where many non-specialists oversimplify the picture.

What Parkinsonian movement looks like
Parkinson’s disease usually produces a hypokinetic movement disorder. The classic features are resting tremor, bradykinesia, and rigidity. The tremor tends to be rhythmic. Bradykinesia is less dramatic to watch, but it is often more disabling. Patients may take extra time to rise from a chair, turn in bed, button clothing, or initiate a first step.
In practice, the gait tells you a lot. You may see reduced arm swing, shortened stride, difficulty turning, and episodes where the patient feels “stuck”. Family members often describe this as stiffness or slowness rather than weakness.
Common observations include:
At rest: a hand tremor that appears when the limb is supported and lessens with purposeful action.
During walking: short, shuffling steps and reduced automatic movement.
During conversation: a quieter voice, decreased facial animation, and longer pauses before movement begins.
What Huntingtonian movement looks like
Huntington’s disease usually produces a more hyperkinetic and less predictable pattern. Chorea is the hallmark. These are involuntary, dance-like movements that seem to flow from one body part to another. They are not rhythmic like tremor. They can look almost purposeful until you watch long enough to realise the person is constantly correcting for movements they never intended to make.
The person may seem unable to sit still, keep the tongue protruded steadily, or maintain a stable posture. Voluntary movement also degrades. Reaching becomes less accurate. Eye movements may be hard to control. Walking may look irregular rather than merely slow.
Families often misread early chorea as anxiety, restlessness, intoxication, or poor concentration. That’s understandable. The motor pattern can masquerade as behaviour.
The quality of instability is different
A balance comparison using the BESTest found that in early-stage patients, those with Parkinson’s disease performed better overall than those with Huntington’s disease, with PD mean scores outperforming HD by a 95% confidence interval of [6.53, 24.18] in this balance study. Clinically, that aligns with what many teams see: early Parkinson’s may preserve more postural organisation than early Huntington’s, where chorea and motor impersistence can destabilise the entire system.
That difference matters during rehabilitation. Programmes built around cueing, pacing, and initiation often fit Parkinson’s well. Patients with Huntington’s may need a different emphasis that accounts for inconsistency, safety risk, and difficulty sustaining controlled movement. Teams working in structured rehab settings can often adapt these strategies through focused neurorehabilitation care pathways.
Bedside clues that help
A side-by-side motor check can be more revealing than a long symptom list.
Bedside task | Parkinson’s disease | Huntington’s disease |
|---|---|---|
Finger tapping | Slow, decrementing amplitude | Irregular, less sustained control |
Rising from chair | Slowed initiation, rigidity | Variable control, extra movements |
Gait | Shuffling, reduced arm swing | Lurching, unpredictable, choreiform intrusions |
Sustained posture | Can hold but with tremor or rigidity | May break posture due to motor impersistence |
Facial expression | Masked facies | Variable, sometimes restless or fragmented movement |
Clinical tip: Ask the patient to perform a simple repeated sequence, then continue talking while they do it. Parkinson’s often looks slower and smaller. Huntington’s often becomes more irregular and less suppressible.
What works diagnostically is not asking, “Is there abnormal movement?” It’s asking, “What kind of abnormal movement is this, and what happens when the person tries to control it?”
Beyond Movement Cognitive and Psychiatric Profiles
The most important diagnostic clues often sit outside the motor exam.
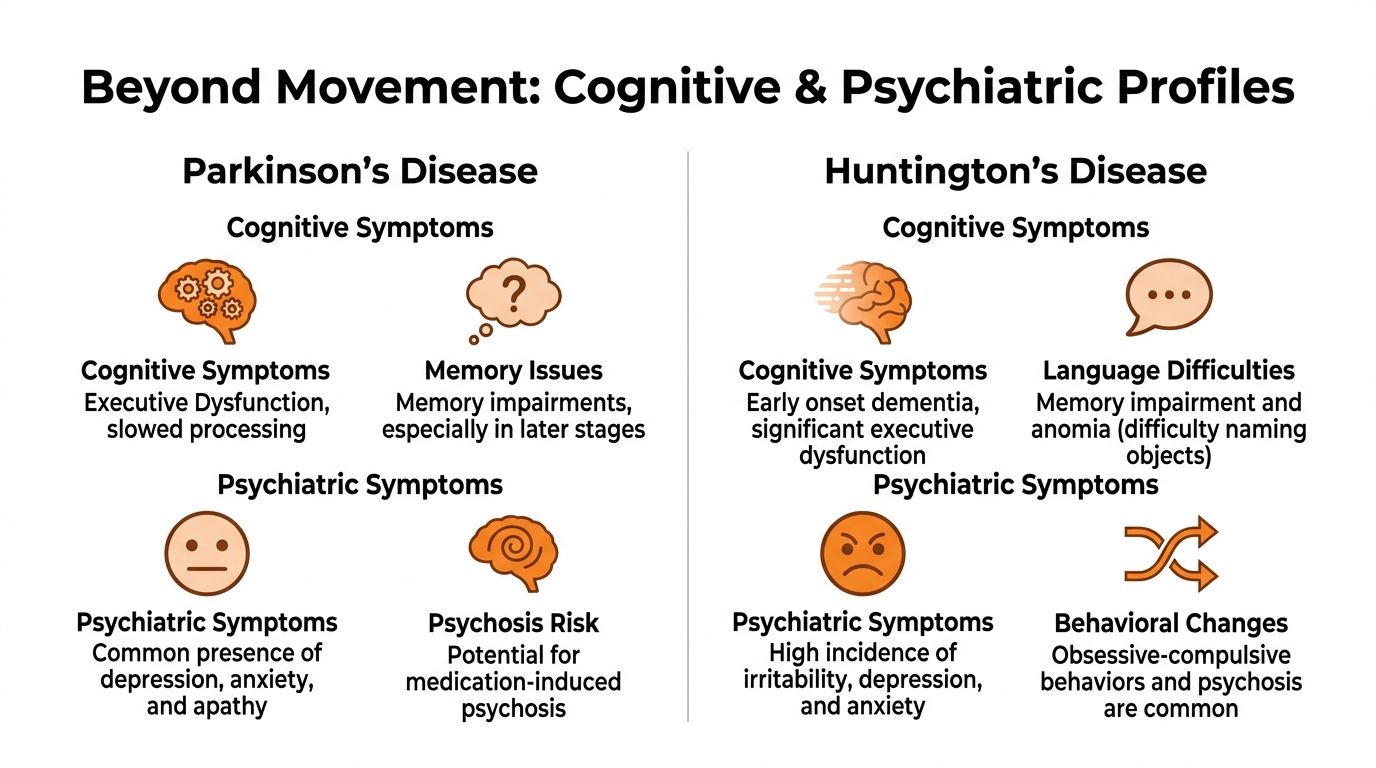
A patient with mild slowness and rigidity may still manage finances, appointments, and medication schedules well. Another patient with relatively subtle involuntary movement may already be making poor decisions, losing work performance, forgetting multi-step tasks, or reacting with unusual anger. That difference often points clinicians in the right direction faster than the movement label alone.
Cognitive pattern in Parkinson’s disease
In Parkinson’s disease, early cognitive difficulty often clusters around attention, executive function, processing speed, and visuospatial ability. Families may say the person is slower to organise, slower to shift set, and less efficient with tasks that require planning under time pressure.
That doesn’t mean memory is normal. It means the earliest pattern often looks more like inefficient retrieval and slowed organisation than a broad primary memory syndrome. Patients may benefit from cueing, structure, and extra time because the bottleneck is often executive.
Typical examples include:
trouble managing two-step or three-step tasks when rushed
slower mental flexibility during conversation
getting lost in complex visual environments such as busy car parks or unfamiliar corridors
difficulty initiating tasks even when they know what needs to be done
Cognitive pattern in Huntington’s disease
A controlled study comparing matched HD and PD groups found that Huntington’s disease patients performed markedly worse across multiple cognitive tests, including general cognition, memory, and language fluency, while Parkinson’s disease deficits were milder and centred more on attention, executive, and visuospatial domains in this cognitive profiling study. Clinically, that’s a major distinction.
In Huntington’s disease, decline often feels broader earlier. The patient may show reduced judgement, poorer self-monitoring, decreased mental flexibility, weakened fluency, and more obvious disruption in daily function. Relatives may notice that the person no longer recognises the impact of their own choices. That loss of insight can become one of the hardest parts of care.
If families need a plain-language frame for these changes, this overview of what cognitive function means in daily life can help translate test findings into practical examples.
Psychiatric symptoms often change the differential
The psychiatric overlap between the two disorders is real. Depression and anxiety can occur in both. But the profile often differs in tone, intensity, and timing.
In Parkinson’s disease, psychiatric symptoms may present as:
Depression or anxiety that accompanies adjustment, neurochemical change, or worsening disability
Apathy that families mistake for laziness
Medication-related psychosis in some cases, especially as treatment becomes more complex
In Huntington’s disease, behavioural and psychiatric change often becomes central much earlier:
Irritability that disrupts relationships or work
Impulsivity that leads to unsafe choices
Apathy that looks deceptively like depression
Obsessive or perseverative behaviour
Psychosis or severe behavioural disorganisation in some patients
When a family says, “The movement changes are there, but the personality change is what scares us,” Huntington’s disease moves much higher on the list.
Why digital cognitive data helps
The practical value of structured cognitive measurement is that it turns vague impressions into trackable patterns. A spouse may report “he’s just slower” or “she’s not herself”. Those are important observations, but they don’t tell you whether the main issue is processing speed, inhibitory control, visual scanning, memory encoding, or behavioural regulation.
Digital tools are especially useful when clinicians need serial data rather than a one-time impression. Short, repeatable cognitive tasks can help answer questions such as:
Is the decline selective and mainly executive, which may fit early Parkinson’s more closely?
Is the profile broader, with language fluency and memory already more affected?
Are psychiatric symptoms overshadowing the cognitive pattern, making baseline quantification even more important?
Is the patient declining steadily enough that care planning needs to change now, not later?
That matters for therapists too. A physiotherapist may see inconsistent carryover. An occupational therapist may notice unsafe sequencing. A speech-language pathologist may detect executive breakdown before formal dementia is obvious. When those observations line up with structured cognitive findings, the diagnostic picture sharpens.
The Diagnostic Journey and Modern Assessment Tools
Diagnosis begins with ordinary clinical work done well. History, examination, collateral information, and pattern recognition still carry most of the weight.

The first pass usually involves listening for the timeline. Did symptoms begin with slowness, tremor, and reduced facial expression, or with irritability, clumsiness, and behavioural change? Did relatives notice a movement syndrome first, or a change in judgement and personality? Is there a known family history that changes the threshold for genetic counselling?
What the clinical work-up should clarify
For suspected Parkinson’s disease, the clinician usually relies on a clinical diagnosis grounded in neurological examination and longitudinal follow-up. The key issue is whether the syndrome behaves like parkinsonism and whether alternative explanations fit better.
For suspected Huntington’s disease, the pathway is different because genetic confirmation can establish the diagnosis. The practical challenge is deciding when behavioural and cognitive red flags are strong enough to trigger referral for counselling and testing.
A sensible diagnostic workflow often includes:
History from both patient and informant because insight may be limited, especially in Huntington’s disease
Motor examination focused on the quality of movement rather than its presence
Psychiatric review because mood, irritability, apathy, and psychosis can distort the picture
Functional review covering work, driving, medication management, and financial decisions
Family history that is explicit, not casual
For families wrestling with the emotional side of hereditary risk, broader reading on genetic testing for mental health can be useful as background. It doesn’t replace disease-specific counselling, but it can prepare people for the kinds of questions that genetic discussions raise.
Where rapid cognitive profiling fits
In California’s diverse population, Parkinson’s disease affects about 170,000 residents, while Huntington’s disease affects roughly 1,000 to 1,500, and misdiagnosis can occur. The same source notes that PD and HD show opposite functional connectivity patterns in the subthalamic nucleus, and that rapid cognitive profiling can help differentiate executive and attention changes in early presentations, especially in underserved groups facing diagnostic delays, as described in this discussion of Parkinson’s versus Huntington’s in California populations.
That doesn’t mean a digital tool makes the diagnosis. It means the tool can capture objective evidence that sharpens referral and monitoring decisions.
Useful domains to assess repeatedly include:
Processing speed, because slowing may be prominent in parkinsonian syndromes
Executive control, especially set shifting, inhibition, and planning
Attention consistency, which often explains real-world errors before a patient scores poorly on broad screens
Eye-hand coordination, where motor-cognitive interaction becomes visible
Memory and fluency, particularly when Huntington’s disease is a concern
A clear explanation of what a neuropsychological assessment involves can help families understand why brief screening, thorough testing, and serial digital measurement each have different roles.
What works and what doesn’t
What works is combining bedside judgement with repeatable data. What doesn’t work is relying on a single dramatic symptom.
A patient with new chorea and strong family history doesn’t need years of vague observation before referral. A patient with subtle bradykinesia and executive slowing shouldn’t be dismissed because tremor is absent.
Another common mistake is overvaluing one normal screening result. Brief screens can miss the earliest executive or behavioural change. A patient may pass a simple global screen yet still be unsafe with medication timing, finances, or driving. That’s why longitudinal profiling often adds more value than a single snapshot.
For therapists and care teams, serial data also improves planning. If the main issue is slowing and initiation, cueing and environmental simplification may help. If the pattern is worsening inhibition, poor insight, and broader decline, supervision and safety planning need to intensify earlier.
Managing the Conditions Treatment and Prognosis
Treatment decisions are where diagnostic accuracy stops being academic.

Parkinson’s disease management
In Parkinson’s disease, treatment often aims to improve function by restoring or mimicking dopamine signalling. That is why dopaminergic medications can be so useful, especially for bradykinesia and rigidity. Rehabilitation also matters. Gait cueing, strength work, balance training, speech therapy, and occupational strategies often improve participation even when medication does the heavy lifting for motor symptoms.
Some patients also benefit from advanced therapies. According to this review of opposing DBS responses in Parkinson’s and Huntington’s disease, deep brain stimulation targeting the subthalamic nucleus can improve motor symptoms in Parkinson’s disease by 40 to 60 percent. That doesn’t make DBS a cure, but it shows how strongly the treatment model in Parkinson’s depends on the underlying circuitry.
Huntington’s disease management
Huntington’s disease requires a different philosophy. There is no equivalent strategy of replacing a missing neurotransmitter and expecting broad motor improvement. Care is largely symptomatic and multidisciplinary. Clinicians may treat chorea, stabilise mood and behaviour, reduce risk, and support communication, swallowing, mobility, and supervision needs as the disease progresses.
The same review notes an important warning: the same DBS approach can worsen chorea in Huntington’s disease, and dopamine-enhancing therapies that are central in Parkinson’s are contraindicated in Huntington’s disease, where dopamine-blocking agents are used to manage chorea. This is one of the clearest examples of why “movement disorder” is not a treatment category on its own.
Treatment trade-offs families should understand
A side-by-side explanation helps prevent confusion.
Question | Parkinson’s disease | Huntington’s disease |
|---|---|---|
Will dopamine-based medication usually help motor symptoms? | Often, yes | No. It may worsen chorea |
Is DBS a standard advanced option in selected cases? | It can be | It may be harmful |
Is psychiatric care optional? | No | No, and often urgent |
Is rehabilitation still useful? | Yes, especially for gait, balance, speech, and ADLs | Yes, but goals often focus more on safety, consistency, and support |
Treatment pearl: If a therapy works because it boosts dopamine, pause before assuming it belongs anywhere near Huntington’s disease.
Prognosis in practical terms
Families usually want more than a disease definition. They want to know how life will change.
In Parkinson’s disease, progression is often long and uneven. People may live with the condition for many years while treatment remains functionally helpful, though falls, freezing, cognitive change, autonomic symptoms, and care complexity often increase over time.
In Huntington’s disease, the course is typically more aggressive in functional terms. Work loss, behavioural disruption, impaired judgement, swallowing issues, and dependency often arrive earlier in adulthood than families expect. The disease affects not only the patient but also relatives who may be caregivers, at-risk family members, or both.
That difference should shape care planning from the start. Parkinson’s often calls for staged adaptation. Huntington’s often demands earlier conversations about supervision, legal planning, caregiver burden, and safety.
Supporting the Ecosystem Caregiver and Family Considerations
Care doesn’t happen only in the clinic. It happens in kitchens, cars, bathrooms, workplaces, and late-night phone calls between exhausted relatives.
For Parkinson’s disease, caregivers often struggle with fluctuating mobility, freezing, falls, slowed thinking, and the growing complexity of medication schedules. Support works best when families simplify routines, reduce environmental hazards, and recognise that slowness is neurological, not oppositional. They also need permission to stop arguing about pace and start building systems.
For Huntington’s disease, the caregiving burden is often heavier emotionally. Families may be managing irritability, poor insight, impulsive behaviour, job loss, and genetic fear across generations. The person needing care may be much younger than the typical neurodegenerative patient, which creates pressure around children, employment, finances, and long-term housing.
Useful steps include:
Create one shared record: keep medication lists, behavioural observations, fall events, and appointment summaries in one place.
Track function, not just symptoms: note changes in judgement, swallowing, driving, finances, and adherence.
Build continuity early: transitions are safer when therapists, neurologists, mental health providers, and family members are working from the same plan. This guide to continuity of care is a practical reference for that process.
Don’t wait for crisis: legal planning, work accommodations, supervision decisions, and family meetings are easier before conflict escalates.
Families usually cope better when they can see change clearly. Objective cognitive and functional tracking often reduces conflict because it gives everyone the same reference point.
The right care plan doesn’t remove grief. It does reduce guesswork, improve safety, and help families make decisions before problems become emergencies.
If you’re trying to clarify a difficult parkinson vs huntington disease presentation, Orange Neurosciences offers rapid cognitive profiling and targeted digital tools that can support referral decisions, longitudinal monitoring, and more personalised care planning. For clinicians, therapists, and families who need clearer data on attention, memory, executive function, processing speed, perception, and eye-hand coordination, it’s a practical next step. You can explore the platform on the website or contact the team by email to discuss how it fits your setting.

Orange Neurosciences' Cognitive Skills Assessments (CSA) are intended as an aid for assessing the cognitive well-being of an individual. In a clinical setting, the CSA results (when interpreted by a qualified healthcare provider) may be used as an aid in determining whether further cognitive evaluation is needed. Orange Neurosciences' brain training programs are designed to promote and encourage overall cognitive health. Orange Neurosciences does not offer any medical diagnosis or treatment of any medical disease or condition. Orange Neurosciences products may also be used for research purposes for any range of cognition-related assessments. If used for research purposes, all use of the product must comply with the appropriate human subjects' procedures as they exist within the researcher's institution and will be the researcher's responsibility. All such human subject protections shall be under the provisions of all applicable sections of the Code of Federal Regulations.



© 2026 by Orange Neurosciences Corporation